台灣醫療器材製造規範
目前臺灣遵循行政院衛生署實施醫療器材優良製造規範(GMP),依據藥事法相關規定、中國國家標準CNS 12681 (ISO 9001)及醫療器材品質保證制度國際標準(ISO 13485) 訂定之。依據臺灣衛生福利部食品藥物管理署訂定之「藥物優良製造準則」第三篇 醫療器材優良製造規範中:
第 18 條:販賣業者應就運銷系統,以書面訂定產品來源及流向之追溯程序;其內容包括產品追溯之範圍及紀錄。前項紀錄,應包括醫療器材名稱、批號、接收日期與數量、保存期限、供應商名稱、收貨人名稱、地址與連絡人、送貨地址、運送方式及允收溫度條件;以包裹運輸寄送者,並應包括收貨人之姓名及收貨地址憑證。第一項之紀錄,應予保存。


第67條:「製造業者應建立與維持紀錄,以提供符合品質管理系統要求與有效運作之證據。品質紀錄應保持清晰易讀、易於鑑別及檢索。製造業者應建立鑑別、儲存、檢索、保護、保存期限及處理品質紀錄之書面管制程序。製造業者保存紀錄之期限應至少相當於製造業者所規定之醫療器材有效期間,且不得少於產品從製造業者放行之日起三年,或按其他相關法規之要求。」
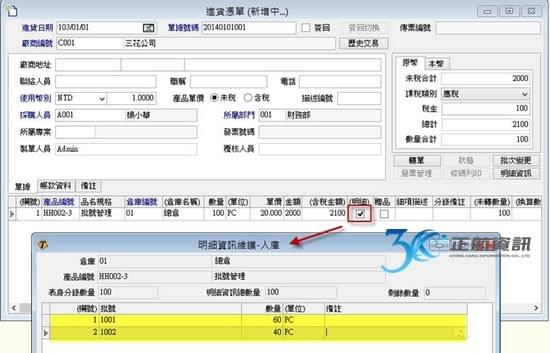
第99條:「製造業者應建立並維持每一批(件)醫療器材之紀錄,以提供本準則所規定追溯性範圍之紀錄,並鑑別生產數量與核准銷售數量。批次紀錄應予以查證與核可。」

法條中也明訂,製造業者應針對每一類型或型號之醫療器材,建立並維持其產品規格及品質管理系統要求之檔案。該文件應具備完整之生產流程,及必要之安裝與服務流程。在此嚴格的規範下,需耗費相當的人力與時間,若能將部分檔案透過電子化傳遞與保存,必要時再將其印出,除能節省檔案傳遞時間外,也可避免在傳遞中遺失。

批次檢驗報告與ERP系統之結合運用
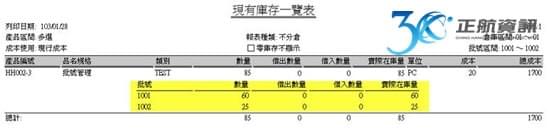
過去,運用ERP系統多只在生產製造中,品質檢驗之過程會記錄在另外的文件中,有可能是手寫記錄或電子檔案留存。當要追朔生產與檢驗之記錄,需另花費相當多的時間來蒐集與整理批次資料。ERP系統導入後之效益,除了最終能提供大量的資料分析與運用之外,最根本之目的在於能詳實記錄完整的作業流程。從進料開始記錄每批原料之進料時間與有效日期,並做到進料檢驗。進入生產流程時,從投料之來源批號,到生產過程所使用之機器設備與設備使用標準等生產歷程皆須落實記錄。生產過程之各工序檢驗需依照不同產成品在不同工序做不同的檢驗項目,特別像是滅菌之重點工序另需做不同的抽查紀錄。除了每批產成品都須有完整的檢驗報告之外,針對每批首件製品也需提供一份檢驗報告。最終,每批產成品銷售至何客戶也須從系統完整追蹤。所以從採購、生產至銷售,再加上批次檢驗報告,若能利用一套ERP系統做緊密之結合,則能大量節省人員須在不同地方記錄與蒐集資訊的時間。

上述提到之各項記錄,為因應製造不同醫療器材所需記錄的資訊不同,在一般模組式ERP只能依賴客製開發來增加記錄欄位,檢驗報表也可能需要重新開發新的格式。若ERP系統能提供工具調整既有單據的欄位與新增全新單據來完整記錄生產歷程,且有報表工具供自訂符合ISO文件之列印格式,便可省去額外的客製費用。